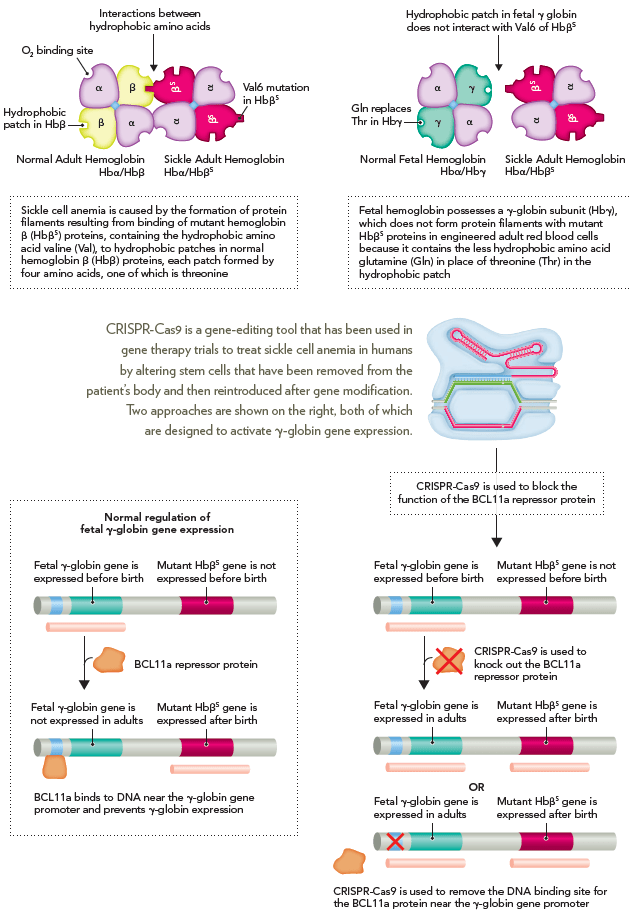
The CRISPR-Cas9 system has recently been applied to the treatment of sickle cell anemia in a small group of patients who suffer from the disease. Sickle cell anemia is caused by a recessive mutation in the human β-globin gene that converts the amino acid glutamate to valine on the surface of the mutant protein as a result of an AT transversion in DNA. This single nucleotide mutation creates a binding site on the HbβS globin protein that leads to the formation of protein filaments caused by the binding of HbβS globin proteins to other Hbβ globin proteins. The fetal γ-globin protein does not contain a hydrophobic patch on the surface that can interact with the valine residue in the HbβS protein. Thus, knocking out the BCL11a gene knocks out the BCL11a repressor protein, which leads to activated expression of the γ-globin gene in adult erythrocyte precursor cells.