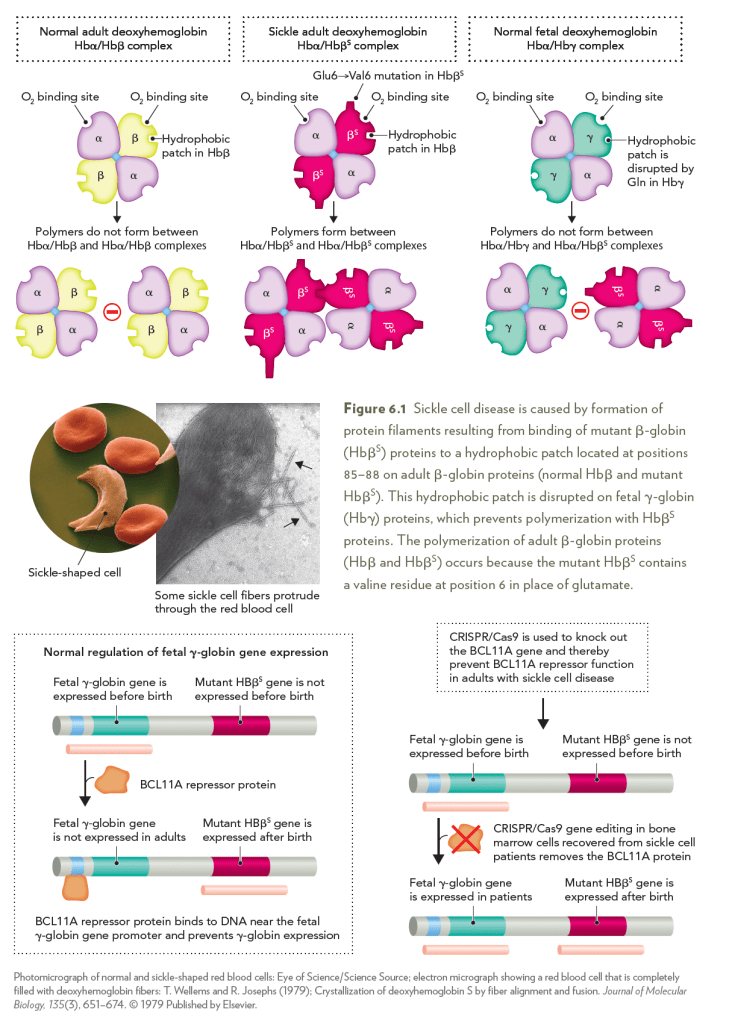
Sickle cell disease is caused by a recessive mutation in the human β-globin
gene that converts the amino acid glutamate to valine at residue 6 (E6V).
The E6V mutation places a hydrophobic amino acid on the surface of
the mutant β-globin (HbβS) protein that interacts with a hydrophobic
patch on adult β-globin proteins. Interactions between hydrophobic
amino acids on HbβS proteins lead to the formation of large hemoglobin
protein polymers that disrupt red blood cell function and cause anemia
in patients with sickle cell disease. It was discovered that the hydrophobic
patch on adult Hbβ proteins is disrupted in fetal γ-globin (Hbγ) proteins,
which prevents HbβS proteins from polymerizing with fetal Hbγ
proteins. Patients with sickle cell disease can be functionally cured of the
disease by reactivating Hbγ gene expression in adult erythrocyte precursor
cells, which is achieved by use of a BCL11A gene knockout strategy
in CRISPR-Cas9 gene editing.
Study guide: Chapter 6 — Protein Function
