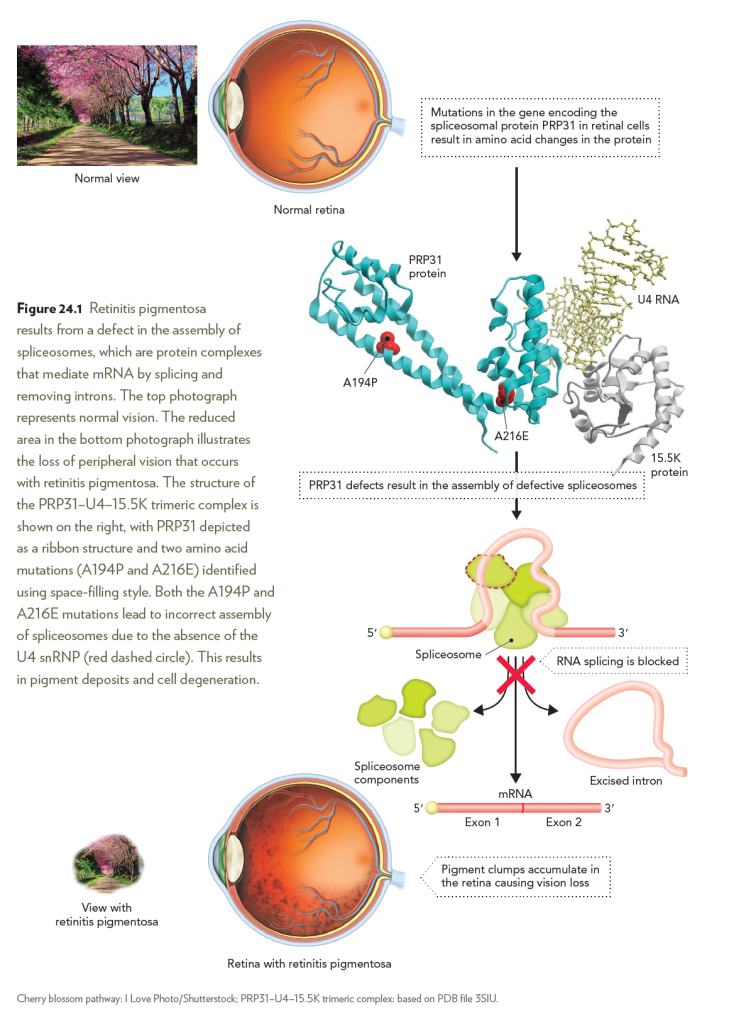
Retinitis pigmentosa is a degenerative disease of the retina characterized
by loss of the light-sensing photoreceptor cells and black mottling
of the retina. Mutations in factors that mediate RNA splicing have been
associated with the development of retinitis pigmentosa. For example,
inherited missense mutations in the gene encoding the human protein
PRP31 result in an autosomal dominant form of the disease. The PRP31
protein promotes assembly of the PRP31–U4–15.5K trimeric complex,
which is essential for correct splicing of mRNA. The two PRP31 missense
mutations, A194P and A216E, are believed to lead to interference
with formation of the trimeric complex, thus preventing proper binding
of the spliceosome to mRNA to facilitate intron removal. Loss of
PRP31-mediated RNA splicing in photoreceptor cells leads to cell death
and vision loss.
Study guide: Chapter 24 — RNA Metabolism
